

Book review: "Innovation Breakdown"
Book review: "Innovation Breakdown"
Subtitle: "How the FDA and Wall Street Cripple Medical Advances"


Innovation Breakdown
By Joseph V. Gulfo
258 pages
2014 Post Hill Press (Amazon link)
Innovation Breakdown tells the story of a small startup with a promising lifesaving product that was unexpectedly screwed over by the FDA. As a result of the FDA’s actions the company’s reputation was permanently harmed, their device took several extra years to make it to market, and many people died who might have been saved.
Innovation Breakdown is a remarkable account. It’s the only account I know of that thoroughly details the process of bringing a new medical device through the FDA. Most remarkably, the author does not hold back his tongue. As Mary Ruwart describes in her book Death by Regulation, biotechnology executives are generally very reluctant to criticize the FDA because they fear the all-too-real possibility of retaliation.
Background on MelaFind
MELA Sciences grew out of an earlier company called Electro-Optical devices. The author of the book, Joseph Gulfo, was asked to be CEO in 2003, partially on the basis of his previous experience with the FDA. By that time scientists and engineers at the company had developed a multispectral camera that images skin lesions using both visible and infrared light. By analyzing the resulting images with machine learning algorithms they planned to identify melanoma, a rare but often fatal type of skin cancer.
Their device, which was christened “MelaFind”, was never intended to replace a dermatologist’s decision making. Rather, it was intended to provide additional information to inform a dermatologist’s decision as to whether to order a biopsy. The device is intended to help dermatologists miss less melanomas when they are faced with tricky cases where it isn’t clear whether to biopsy or not.
Most skin cancers are basal cell carcinomas, which are rarely fatal. About 1% of skin cancers are melanomas. Melanomas kills 8,000 people annually in the US, about one person per hour. It is the number one killer of women between age 30 and 35. Early detection is critical for melanoma. Melanomas that are caught early when they penetrate less than a mm into the skin have a 15 year survival rate of 93%. If the cancer grows to penetrate deeper than one mm, however, that survival rate drops to 68%.
Biopsies are expensive, invasive, and often painful procedures, so biopsying everything is not a good option. Dermatologists often struggle with the decision of whether to biopsy or not. Prior to Melafind, on average only 1 in 30 biopsies tested positive for melanoma. Melafind helps target the biopsies better, resulting in less melanomas being missed while also reducing the number of biopsies performed overall.
At the time MelaFind was invented many doctors were already using an algorithm to help them decide whether to biopsy. That algorithm is called the “ABCDE criteria”. ABCDE refer to five possible attributes that a lesion may have - Asymmetry, Border irregularity, two or more Colors, Diameter greater than 6 mm, and Evolving. According to that criteria, a lesion that demonstrates all five features should be biopsied. I mention this just because I find it amusing that doctors can invent algorithms and use them without any FDA approval. Doctors can even sell these algorithms in textbooks and medical school courses. If you want to sell a computer-based algorithm, though, the FDA has to be involved.
Background: how medical device approvals work
There are two paths for medical device approval at the FDA - PMA (Premarket Approval) and 510k. The 510k pathway is the easiest pathway and the way most medical devices are approved. To qualify for 510k, the applicant must demonstrate “physically equivalent properties” to a previously approved device. The PMA is for “first in class” devices such as MelaFind. For the 510k the FDA is required to render a decision within 90 days, and for the PMA they have 180 days. However, there are many reasons the FDA can stop and start this “regulatory clock”. For instance, any time the FDA asks the company a question they are allowed to stop the clock. So, in practice the 510k process typically takes 6-12 months while PMA process takes 2-3 years. The FDA charges companies about $20,000 for the 510k process and $440,000 for PMA. Prior to application, the company and the FDA work together to design a clinical trial. Once the company has submitted their application and all their data, a panel of independent experts is convened to vote on the device. The panel votes on three questions that are centered on safety, effectiveness, and benefit/risk. The panel’s votes are taken into consideration by the FDA but they do not bind their action. Assuming the panel vote is favorable the FDA then enters into negotiations with the company about the wording of the label, a process that can take several months. Once the label has been decided the device will be approved (or not). As demonstrated in the MELA case, this process does not always go as it is supposed to, which creates regulatory uncertainty and lowers investor’s appetite to invest in novel biotechnology.
First contact
Some people think that the way FDA approval works is that the company does a trial, writes up the results, and submits an application. In actuality, companies must engage in a continual dialogue with the FDA starting long before they start any trials. The FDA is always very particular about what sort of trials they would like to see run, and they are known to change their minds mid-flight (the problem of “moving the goalposts”).
When Gulfo became CEO the company had little relationship with the FDA. Previously, employees had only talked with the FDA’s Office of Science & Technology (OST), which is charged with keeping the FDA abreast of emerging technologies. The people there were excited about the device. Unfortunately that enthusiasm didn’t count for much because the OST doesn’t actually conduct any reviews. Gulfo set out to establish a rapport with the FDA’s reviewers in January 2004. He started with a phone call but got no response. He then wrote a letter. Again, no response. He sent multiple emails to different review divisions. No response. Finally he got a meeting. According to Gulfo, the FDA always gives a reflexive response of “No way - shut down your company now” during first meetings. With persistence, though, it’s possible to convince the FDA you may have something worthy of approval. In the case of MelaFind by the end of their second meeting the FDA was onboard.
On October 20th, 2004 MELA Sciences received a binding protocol agreement with the FDA. This agreement is similar to the way pre-registration works at some journals. Under such an agreement the FDA agrees in advance on the sort of clinical trial and endpoint criteria necessary for approval. Such agreements are highly sought after but rarely given out by the FDA, because they bind the FDA in advance to having to make a decision if certain criteria are met. The FDA gives out just one or two binding protocol agreements a year, at most. Furthermore, because MelaFind was a new device satisfying an unmet need, the FDA deemed MelaFind a “breakthrough device” and granted MELA “expedited review” status.
Disaster
Mela’s trial ran between 2007 - 2009 and was the largest study ever conducted in dermatology. They found that their device had a sensitivity (true positive rate) of 98.3% compared to 78% for dermatologists. Furthermore, MelaFind correctly identified 6% more benign lesions as truly benign compared to dermatologists, which would translate to 6% less unnecessary biopsies when in clinical use.
The FDA was very impressed with the results of the study. On November 23, 2009 MELA received an email with this message: “It looks like we are heading towards a March date for the panel.” Gulfo asked if the FDA might be willing to a joint announcement of the device’s approval on May 3rd, which is known as Melanoma Monday (an awareness day run by the American College of Dermatology as part of “Skin Cancer Awareness Month” in May). The FDA was very receptive to the idea. All signs pointed to MELA being on a glide path to approval.
Then, out of the blue, disaster struck. First, there was some radio silence from the FDA. Then, on March 19th, 2010 MELA received a letter in the mail which “exploded like a bomb” in their office. The letter proclaimed that MELA’s device was “not approachable”. It was an official FDA “action letter”, something that is not supposed to come until after the panel. By law MELA was entitled to a panel meeting before such a letter. The FDA had broken the law.
For the next few months, MELA waited for the FDA to retract the letter. New rules for how panels were supposed to be run were coming into effect and Gulfo assumed that the FDA had sent the letter to buy time and delay the panel until after the new rules went into place.
It became clear however that something much more nefarious was at play — the FDA was trying to sink MelaFind. At a meeting on August 26th 2010 the FDA agreed to hold a panel but demanded that the panel vote on an indication that MelaFind could be used by all physicians. This was contrary to what the company wanted - MELA has had always said that the indication should be for dermatologists only. Setting the proposed indication to all physicians would make it much harder to get approved. The only explanation for this action is that the FDA wanted to torpedo the approval at the panel stage.
Prior to the panel, the FDA sent a bunch of materials to the review panel to read beforehand. The FDA is not supposed to prejudge anything in these materials, but the materials included this statement: “The FDA review team has significant concerns that this device has not been studied adequately for its current indications for use and therefore puts the health of the public at risk”. The document also contained numerous pieces of erroneous information that all cast doubt on the device.
The team knew that the FDA was pulling out every dirty trick they could to sink things during the panel, and they did. The team pulled many long hours preparing for the presentation. They had to become antagonistic during the panel meeting, calling out the FDA’s lies. Miraculously, despite the FDA’s numerous attempts to sink the approval, the panel voted in favor of the device by a slim margin of one vote. Most of the panelists who voted no said they would have voted yes if the indication had been limited to use by dermatologists only.
This was an enormous victory for MELA but their struggle with the FDA wasn’t over. For months the FDA refused to engage with the company. The future of the company was in limbo.
MELA goes on the offensive
What happened next is fascinating, at least for those interested in “theory of change”. The company assembled a legal and PR team in Washington, D.C. to go on the offensive. Their core case against the FDA was simple - the FDA had broken the law, not once but twice. First, they issued a “not approvable letter” before the panel was held. Secondly, they had reneged on the binding protocol agreement.
MELA employees began writing letters and holding “desk side briefings” with reporters. They networked like crazy, talking with policy makers and well-connected individuals. Partially as a result of these efforts on March 30, 2011 the Wall Street Journal published an editorial on MelaFind entitled “Medical Progress, Please: Your Next Melanoma, Courtesy of the FDA”.
As part of these efforts Gulfo went to talk with Dr. Michael Mandel, a well-known economist at the Progressive Policy Institute who specializes in matters of regulation and innovation. Dr. Mandel said he had been looking for a CEO brave enough to speak openly about their difficulties with the FDA. Gulfo was more than willing to do so and sent Dr. Mandel all the documents he had regarding the pending FDA approval. Dr. Mandel read all of them and even did his own statistical analyses of MELA’s data. In June 2011 Mandel published a whitepaper entitled “How the FDA Impedes Innovation: A Case Study in Overregulation”.
Around this same time many venture capitalists were starting to speak out as well. MELA wasn’t the only company facing fierce and unexpected opposition from the FDA at this time. Johnson & Johnson was facing a similar situation to MELA with regards to the approval of their Sedasys device for monitoring and administering anesthesia. J&J had done everything the FDA had asked but the FDA kept twisting misrepresenting and twisting the results of their study. Sedasys would be later be rejected in 2010 but ultimately would be approved on appeal in 2013.
The stalled approvals at the FDA created regulatory uncertainty which greatly dampened VC appetite for investing in medical technology startups. The damage of this is hard to calculate but is very clearly massive. An article from June 2011 in Venture Wire identified nine venture capital firms who had complained publicly about the FDA and also identified four companies that had recently shut down due to “the daunting FDA process”.
All of this outcry culminated in a congressional hearing on July 20, 2011. Asked to testify was Dr. Michael Mandel and five other people who had serious complaints about medical device approvals at the FDA. One of the congresspeople who asked questions, Rep. Brian Bilbray, had a daughter who had gotten advanced melanoma because it had gone undetected. FDA Center director Dr. Shuren was also asked to testify. According to Gulfo, Dr. Shuren lied several times during his testimony. That is a pretty serious accusation!
CE mark vs the FDA
The contrast with the “CE mark” process in the EU is quite amazing. According to Gulfo, the CE process was much simpler. They submitted their materials in May 2011 and received approval five moths later on September 6th. For comparison, they submitted to the FDA in June 2009 and ultimately didn’t get approval until November 2011 - 30 months or six times longer. Gulfo says that the CE process does not focus on efficacy as much as the FDA process but is stricter in terms of engineering and safety requirements.
Obviously a reciprocity arrangement between the EU CE Mark organizations and the FDA would make medical device innovation much less burdensome and greatly speed time to market. At the very least people should be able to buy CE Mark devices in the US (with a label clearly stating its CE approved and not FDA approved).
The FDA could also adopt the EU’s system. Alex Tabarrok writes:
“One key reason for Europe's efficient approval process is that European governments don't review medical devices directly. Instead they certify independent "notified bodies" that specialize and compete to review new products. The European system works more quickly than the U.S. system, and there is no evidence that it results in reduced patient safety”
Gulfo’s beef with Wall Street
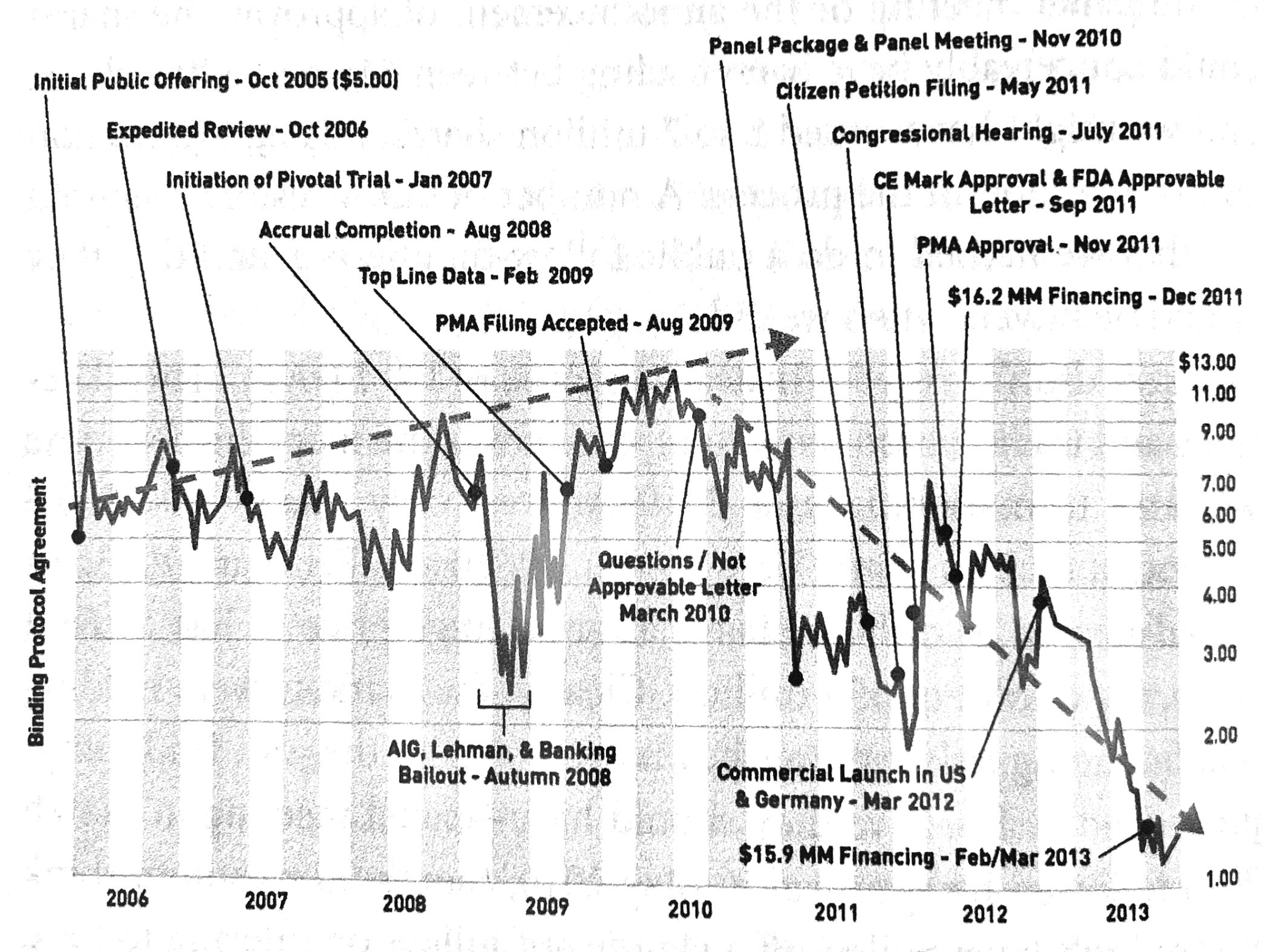
The last 1/3 of the book is Gulfo railing against Wall Street. Gulfo’s complaints about Wall Street don’t land quite as strongly as his complaints about the FDA. Gulfo admits they took MELA public too early and many of the problems they encountered most likely would have been avoided if they had not done so. Furthermore, MELA’s problems with Wall Street all began with the FDA’s bombshell “not approvable” letter. I’m not sure its reasonable to expect Wall Street investors to react a lot differently then they did. Gulfo believes that investors, especially long-term fundamental investors, should have realized that the letter was unjustified and illegal. However, from a Bayesian “outside view” perspective, selling the stock upon seeing such a letter to a small one-product startup makes sense — normally such an action would mean greatly increased probability that the startup will go bankrupt. Furthermore, Gulfo admits that even after they finally got FDA approval they had trouble selling their product because the reputation of MelaFind in the dermatology community had been severely damaged by the FDA’s actions and statements. So, the Wall Street investors who sold after the FDA’s letter appear to be have been right in their assessment — the company ended up much profitable than it could have been long term.
Up until that letter, MELA’s stock had been rising, even through the 2008 financial crisis. After the FDA’s letter many fundamental and venture capital investors sold their positions and activity on the stock became dominated by swing traders and short sellers. MELA’s stock continued to fall for years after the FDA finally approved their device and they were shipping product successfully. According to Gulfo, the short sellers spread FUD (fear, uncertainty, and doubt) on places like Yahoo message boards. Additionally, once the stock started falling, several negative feedback loops set in. For instance, many funds have a minimum investment size and a maximum amount of a company they can own hard-coded into their charter. So, if a fund’s minimum investment is $5M and their max percent ownership is 2.5%, they cannot invest in any company with a market capitalization less than $200M. As MELA’s market cap fell, the number of funds willing to invest them also decreased. The falling stock price also made it much harder to do fundraising rounds. Gulfo complains that they had to do 11 fundraising rounds to keep the company afloat and that arranging all those rounds took enormous amounts of time and effort.
Gulfo’s main beef with Wall Street is that there are too few fundamental investors with a long-term outlook. Gulfo also complains that Wall Street investors and bankers only care about making money and don’t care at all about how a product will benefit patients. He contrasts this with the venture capital and private investors who invested in MELA early on who he says did care about helping patients. I am sympathetic to Gulfo and I think it would be nice if there were more fundamental investors on Wall Street with a deep knowledge of biotech as well as more investors who cared about helping patients. Overall, however, I think his case against the FDA is stronger, and the necessary fixes to improve the FDA are much clearer.
Is Dr. Jeff Shuren to blame?
At interesting part of the account is how little insight Gulfo was able to get into what went wrong at the FDA. The FDA’s actions against MELA make no sense from the outside. We are left only to theorize. One clue is that on January 2010 Dr. Jeffrey E. Shuren became head of the FDA’s Center for Devices and Radiological Health, a position he continues to hold to this day. The “not approvable” letter came just three months later, leading to speculation by Gulfo that Shuren issued a directive to torpedo the approval.
Interestingly, in 2020 - 2021 Shuren as instrumental in making sure that Americans did not have access to Covid tests during the pandemic. Dr. Shuren appears to be a regulatory ideologue - someone who thinks the FDA made must be made as strict as possible to “protect” people.
According to Gulfo, this is a man who was willing to lie under oath to protect the reputation of the FDA.
Conclusion
MELA’s story is eye-opening in many ways. It raises the question — how can we prevent what happened to MELA happening again? How can we speed approvals, reduce regulatory uncertainty and increase transparency? The last chapter of the book contains Gulfo’s “Medical Innovation Manifesto” - a surprisingly granular list of specific policy proposals. I found it very dry reading, but I have little doubt that if all his proposals were implemented together it vastly improve things at the FDA.
Gulfo isn’t proposing that we “deregulate all the things” and I don’t think that is the right path either. Some aspects of the FDA are too strict, others too loose. Certainly there is a general trend towards over-regulation, driven by fear, but over-regulation is not always the problem across the board.
The overarching problems Gulfo describes are due to mission creep and fear. FDA’s legal mandate says that the role of the agency is to “promote the public health by promptly and efficiently reviewing clinical research and taking appropriate action on the marketing of regulated products in a timely manner”. As Gulfo points out, everywhere on their website and materials the FDA describes their role as to protect the public health rather than promote it. What appears to be a minor shift in wording actually is indicative of a major underlying shift in philosophy. The law was written so that the FDA should measure safety to determine indication for use. All medical devices and drugs have risks. The FDA’s role is to make sure the risk are well quantified and to delinate the situations (the indication) where the risk is worth taking. Ultimately we want doctors to decide on when risks are work taking, on a case by case basis.
Instead the FDA, out of fear, has shifted towards protecting people from any possible harm by blocking access to drugs and products. This leads to the invisible graveyard. According to Gulfo, about one person per hour dies of a skin disease that is visible on the skin. From the FDA’s perverted point of view though this state of affairs is safe, however. The FDA generally only focuses on potential harm from companies looking to improve the status quo and not the harm of leaving the status quo as is.
Gulfo also describes substantial scope creep at the FDA. The intention of the law, according to Gulfo, was that the FDA does basic effectiveness testing - does something help, or not? Today however, the FDA factors in relative performance in its approval and is demanding more and more burdensome efficacy testing on larger and larger populations over longer periods, which stifles innovation.
There is room for debate about how much effectiveness testing is needed. In some areas, such as cancer drugs, the effectiveness standards are probably too low (leading to what Vinay Prasad calls “medical reversal”. In medical devices, though, the standards appear to be too high, and this is significantly dampening investment and innovation.
There are also areas where there is utter lack regulatory clarity. Gulfo says that nanotech is one such area — the FDA has no idea how to regulate nanotech, which is stifling investment. As I’ll describe in a future post, a similar situation is happening now with general purpose medical AI.
Further reading
Review in the Wall Street Journal by Alex Tabarrok
Wow ... deserves a lot more attention. Thanks for posting
I found this very insightful! By now, I have heard people rage about the FDA so many times but still don’t have great clarity on what exactly makes (parts of it) so terrible.
Also I would like to learn more about the differences between Europe and the US in device and drug approval processes.
You have written about the FDA multiple times. What makes you personally so interested in this topic?